- Published on
To quantify the AI-boosted materials discovery
Many recent reports discuss machine learning (ML) accelerated approaches to discovering materials and managing energy systems. As a result of this research effort, we posit that there should be a consistent baseline from which these reports can be compared.
The primary goal in materials discovery is to develop efficient materials that are ready for commercialization. The commercialization of a new material takes intensive research efforts that can span up to two decades: the goal of every accelerated approach should be to accomplish this in an order of magnitude less time. The materials science field can benefit from studying an analogous case of vaccine development. New vaccines historically take 10 years from conception to market. However, in less than one year from the start of the COVID-19 pandemic, several companies were able to develop and begin releasing vaccines. This achievement was in part due to an unprecedented global research intensity, but also by a shift in the technology: DNA-sequencing underwent a paradigm shift in 2008, and the cost of sequencing DNA began decreasing exponentially, significantly faster than Moore’s Law, enabling researchers to screen orders-of-magnitude more vaccines than was previously possible.
ML for energy technologies has many commonalities with other fields like biomedicine. They both are the extensive application scenarios of ML advances from the computer algorithms field, sharing the same methodology and principles. However, the difference does exist when talking about the practice of employing ML in different fields. Attacking distinct problems may expose the model to extra unique requirements. For example, ML models for medical applications have to build a complex structure to enable regulatory oversight to ensure the safe development, use, and monitoring of systems, which usually won’t happen to the energy field. Meanwhile, data availability varies significantly from field to field that biomedical researchers can work with a relatively large amount of well-accumulated data which energy people usually lack. And the limited accessibility to a sufficiently large amount of data could constrain the usage of sophisticated ML models that can have more capabilities (such as deep learning models). However, adaption has been rather quick among all fields with a rapidly increased number of groups recognizing the importance of statistical methods and starting to use them for various problems. We posit that the use of high-throughput experimentation (HTE) and ML in materials discovery workflows can result in a similar paradigm shift but will first need a set of metrics by which they can be evaluated and compared, so that they may better improve.

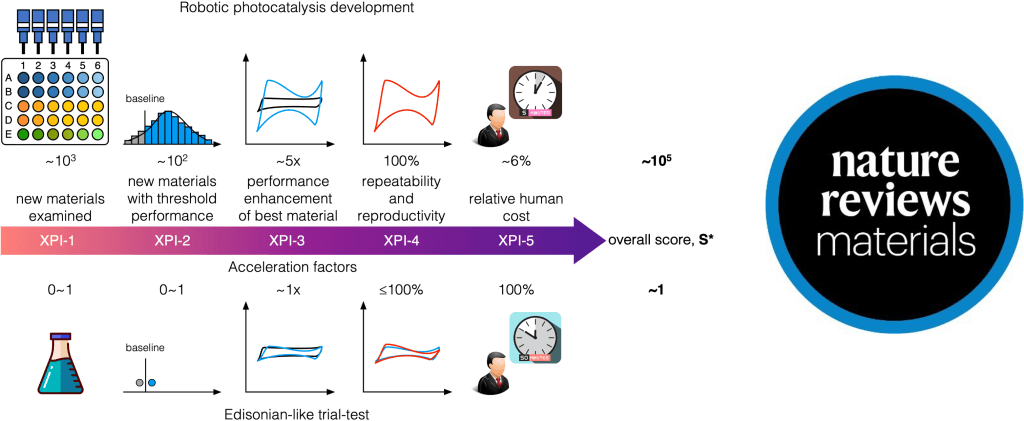
Accelerated materials discovery methods should be judged on the time it takes for a new material to be commercialized. We recognize that this is not a useful metric for new platforms, nor is it one that can be used to quickly decide which platform is best suited for a particular scenario. To this point, we propose here Acc(X)eleration Performance Indicators (XPIs) that new materials discovery platforms should report.
Acceleration factor (AF) of new materials
This XPI will be evaluated by dividing the number of new materials that are synthesized and characterized per unit time with the accelerated platform by the number of materials that are synthesized and characterized with traditional methods. For example, an AF of 10 means that for a given time period, the accelerated platform can evaluate 10x more materials than a traditional platform. For materials with multiple target properties, researchers should report the rate-limiting AF.
Number of new materials with threshold performance
This XPI tracks the number of new materials discovered with an accelerated platform that have a performance greater than baseline value. The selection of this baseline value is critical – it should be something that fairly captures the standard to which new materials need to be compared. As an example, an accelerated platform that seeks to discover new perovskite solar cell materials should track the number of devices made with new materials that have a better performance than the current record solar cell.
Performance of best material over time
This XPI tracks the absolute performance – whether it is Faradaic efficiency, power conversion efficiency, or other – of the best material as a function of time. For the accelerated framework, this should follow a trajectory which grows more rapidly than the counterpart of the traditional methods.
Repeatability and reproducibility of new materials
This XPI seeks to ensure that the new materials discovered are consistent and repeatable – this is a key consideration for commercialization and can be used to screen out materials that otherwise would only fail at the commercialization stage. The performance of a new material should not vary by more than x% of its mean value: if it does, this material should not be included in either XPI-2 or XPI-3.
Human cost of the accelerated platform
This XPI reports the total costs of the accelerated platform. This should include the total number of researcher hours that were needed to: design and order the components for the accelerated system; develop the programming and robotic infrastructure; develop and maintain databases used in the system; and maintain and run the accelerated platform. This metric will provide researchers with a realistic estimate of the resources required to adapt an accelerated platform for their own research.
Each of these XPIs can be measured for computational, experimental, or integrated accelerated systems. Consistently reporting each of these XPIs as new accelerated platforms are developed will better allow researchers to evaluate the growth of these platforms and can provide a consistent metric by which different platforms can be compared. As a demonstration, we applied the XPIs to evaluate the acceleration performance of several typical platforms: Edisonian-like trial-test, Robotic photocatalysis development, and DNA-encoded library-based kinase inhibitor design. As the reference, the Edisonian-like approach has a calculated overall XPIs score around 1, while the most advanced method among them, the DNA-encoded library-based drug design, exhibits a score/acceleration factor of 107. For the sustainability field, the Robotic photocatalysis platform shows an overall XPIs score of 105, catching up with the biological counterpart.







